


Developing biochemistry and algorithms for a computer-aided DNA design based on DNA reuse (Leader WEIZMANN)
Task 2.2: Algorithms for planning the DNA processing and library construction (Leader: WEIZMANN) (M01 – M36)
Partners Weizmann and UNOTT developed an algorithm for planning multi-stage assembly of DNA libraries with shared intermediates that greedily attempts to maximize DNA reuse. As such it improves CADMAD's core technological feature, which is DNA reuse and combinatorial assembly. A manuscript describing this work has been published in ACS synthetic biology (UNOTT and Weizmann) [ACS Synth Biol. 2014 Aug 15;3(8):529-42. doi: 10.1021/sb400161v. Epub 2014 Apr 14.]. Below is a brief description of the work performed during the past year in developing new and improved algorithms for DNA editing:
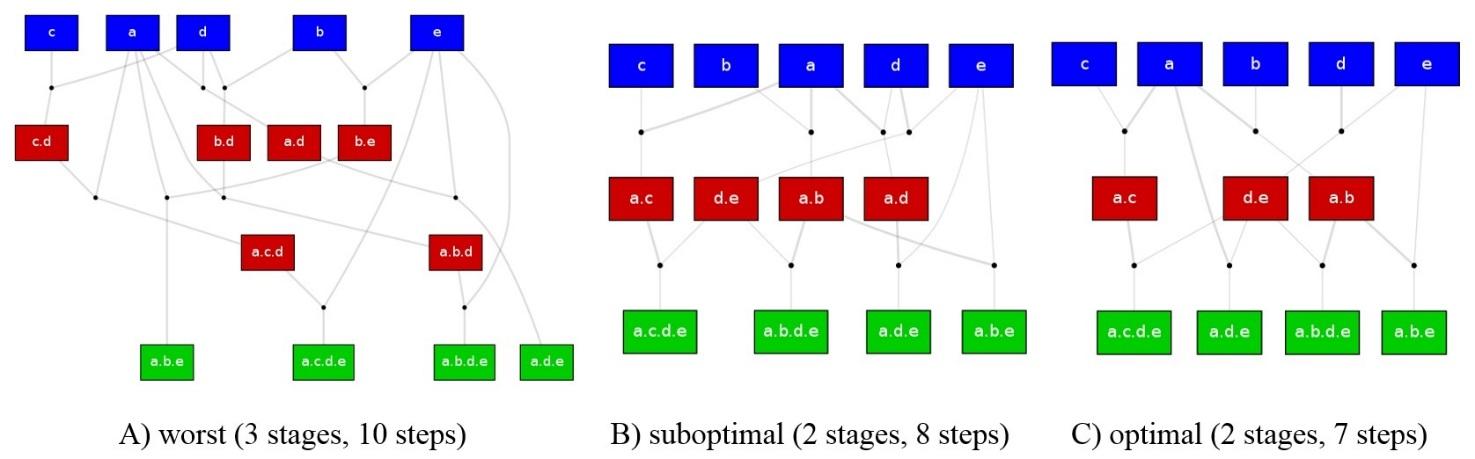
A DNA library assembly task includes a set of input molecules, a set of output molecules (targets) and assumes a binary part concatenation operation that can select two parts within existing (input or intermediate) molecules to produce their concatenation. An assembly plan that produces the outputs from the inputs can be viewed as a graph representing the binary part concatenation operations leading to the set of targets from the set of inputs through a number of intermediates. Figure 2.2 visualizes three possible plans for constructing a simple library composed of 4 targets. The quality of a plan can be categorized by the pair of integers (stages, steps), which allows to compare the outputs of assembly planning algorithms. A plan is considered to be better if it has fewer stages and fewer steps. There may be a trade-off between stages and steps, however whether a suboptimal number of stages with significantly fewer steps is preferable or not is a technology-dependent manufacturing decision.
Starting from each library’s available unitary parts, the algorithm repeatedly - in analogy to independent steps within an assembly stage - concatenates a subset of the remaining pairs of adjacent parts within the sequences, until no pairs remain, meaning every target has been successfully reassembled. The sets of pairs concatenated at each stage of the execution thereby constitute a viable assembly plan.
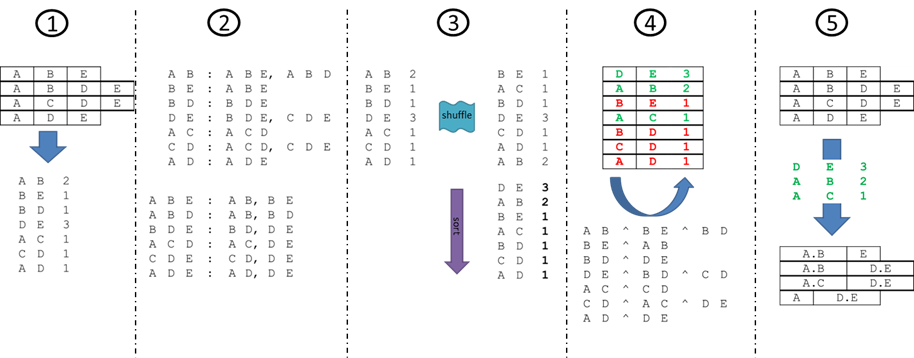

While targets remain to be assembled, each iteration progresses in five phases:
- Occurrences of every pair are counted;
- Pairs are mapped to containing triplets and vice versa;
- A list of pair-count tuples is shuffled (blue wave) and stably sorted by count descending (purple arrow);
- For each yet-to-be-excluded pair, choose pair to be a step (green rows) then exclude other pairs (red rows) sharing the same containing triplets using mappings from the second phase; finally
- Concatenate all instances of each step-pair.
Note that in this example, no triplets exist in the second iteration, as the structure of the library after the first iteration is exactly the set of pairs that will comprise the steps of this final stage.
In the paper, we showed that the abstract computational problem bounded-depth min-cost string production (BDMSP) underlying efficient combinatorial DNA library assembly is NP-hard and APX-hard (meaning it is unreasonable to devise an optimal algorithm to the problem within feasible run time). Hence, in its current form, our method does not guarantee optimal solutions to the very difficult problem of devising assembly plans. Nevertheless, through a combination of greedy heuristics and randomization, our new algorithm consistently produces a tight distribution of high quality plans, and can therefore be run repeatedly to quickly generate a set of solutions that can be filtered or augmented according to additional criteria relevant to automated manufacturing of these large combinatorial DNA libraries. From both design and production perspectives, it can give quick and reliable feedback, often in real-time, as to number of necessary assembly steps and their identities, a proxy for the effort and costs involved.
The presented simple pairs ranking by count can be expanded to more complex pair scoring functions. Taking into account various production criteria such as biochemical ease of assembly (fragment length, low complexity regions, etc.), regional robustness in downstream processing or presence in longer repeated elements (trios, quads etc.) calls for a multifragment assembly. Other considerations such as mounting input fragments on primers in the final assembly process or grouping multiple binary assemblies into a single n-ary assembly can be added as a pre- or post-process if desirable.
To utilize our published algorithm in the library production workflow, exactly these kind of changes needed to be implemented. During the last period we have revised the published algorithm's code so that more complex scoring functions can be taken into account, considering the rapidly updating oligo synthesis capabilities and production technologies.
The main fundamental change is the ability to neglect the costs of assembling fragments who's assembled product can be synthesized from scratch more efficiently. Assembly costs easily outweigh de novo synthesis since assembly requires the synthesis of fragments (de move synthesis of oligos) plus the costs of lab work, reagents, and the possibility to accumulate errors. The availablity and costs of modern product such as minigenes, gblocks and ultrameres, continually tilt the balance further towards synthesis of larger bits of the library.
That said, the number of synthetic part required for assembling a set of sequencing that http://www.viagrabelgiquefr.com/ contains multiple combinatorial features grows exponentially as the synthetic fargments get longer and are there for mounted with more combinatorial features and with it the costs. It is that delicate balance that is being addressed by this expansion of the scoring considerations in this revision of our published algorithm.